
A Mechanist's Guide to the Coronavirus Genome
Hello and welcome to my Coronavirus Genome Walkthrough.
(Hoping someone comes out with that Vaccine Speedrun soon. This boss battle is really shaping up to be an intense one and we’ll need all the artifacts we can get.)
Here, I aim to provide a mechanistic explanation of the SARS-CoV-2 genome’s syntax and semantics. Let’s investigate what the SARS-CoV-2 viral genome actually does as if reading through code like a compiler, from nucleotides to amino acids all the way to proteins. From the four base pairs all the way up to the completed protein-coated virus, what is a virus like this is actually made of on the concrete, physical level?
Understanding a Full System
The underlying purpose of this essay is less about the coronavirus per se and more about how having a small—but functionally complete—piece of viral RNA to analyze gives me a unique opportunity to try to understand a complete self-replicating machine from scratch. This is not a feat that I would have the fortitude to manually replicate with the full human genome, for example—but the coronavirus genome, like the nematode genome, is small enough that we stand a chance at building a complete understanding. The task is perhaps akin to interpretability, but for biological systems instead of artificial neural networks.
As a consequence, this essay is not intended to produce epidemiological conclusions; there are plenty of other sources for that! This essay is about fully understanding a biological system at the chemical and physical level.
Play, Curiosity, and Mechanical Understanding
Throughout this essay, I follow my curiosity in the style of serious play: if I notice I’m confused about something, I look into it and explore it until I’m satisfied that I now understand, and that my understanding is a mechanical understanding. Things are made of stuff! It turns out that we can understand that stuff!
I may skip over some details that were not confusing to me during my own research, but your journey need not be the same as mine. If you’re confused about something while reading this essay, I encourage you to go and look it up! Notice when your curiosity arises; that’s the meditation. It’s always possible to discover the frontier of your own knowledge and to expand it.
This all, at least, has been my intention as I set out to create this piece! As Ken Liu said of his philosophy while translating The Three-Body Problem, “I may not have succeeded, but these were the standards I had in mind as I set about my task.”
Part 1, here, covers just the genome and its translation to proteins. I hope to also write a Part 2 which would cover the structure and function of those proteins, their protein-protein interactions, and the full viral life cycle.
Let’s get started.
Viruses
As a reminder, SARS-CoV-2 is a positive-sense single-stranded RNA virus.

What does this mean we can expect?
- Single-stranded: Its genome is a single strand of RNA (ssRNA).
- Positive-sense: That single strand of RNA can be immediately translated into protein by the ribosomes of the cell it infects.
From this we can also infer that one of the proteins the virus encodes for must be RNA-dependent RNA polymerase (RdRP), a protein which synthesizes new RNA given an RNA template. That’s right: RNA → RNA. However, according to the central dogma of molecular biology, isn’t RNA → RNA an unconscionable heresy? Correspondingly, RdRP is not naturally found in cells! All known positive-sense ssRNA viruses therefore must encode RdRP in order to successfully commit this heresy.
…Wait a minute, the phrase “positive-sense ssRNA virus” implies the existence of negative-sense viruses. If those don’t encode their proteins directly, how can they possibly work?
Positive sense and negative sense
Negative-sense ssRNA viruses also exist! Influenza, Ebola, and measles are examples.

The inner contents of negative-sense ssRNA viruses consist not of an RNA genome but of a ribonucleoprotein, which incorporates both an RNA genome as well as a cohort of viral proteins capable of replicating RNA. Unlike positive-sense ssRNA viruses, negative-sense ssRNA viruses must travel with a working copy of their RNA-replicating proteins. This ribonucleoprotein has enzymatic activity!
RdRP as drug target
Since RdRP has (as far as I know) no legitimate purpose in human cells and is not naturally coded by them, might it offer a potential target for novel antiviral drugs?
Velkov et al. 2014 explores RdRP as a drug target for antivirals against the Hendra virus, a negative-sense ssRNA virus, though I am unable to find the full text.
This review examines the current knowledge based on the multi-domain architecture of the Hendra RdRP and highlights which essential domain functions represent tangible targets for drug development against this deadly disease.
There must be some reason that developing antivirals against this protein is technically (or socially) complicated, or I’d have expected us to do it by now – there are a lot of RNA viruses that this drug target could theoretically hit. Flagging this discrepancy for further research.1
The full genome
Back to SARS-CoV-2! First, let’s get us a genome. Obviously this virus has seen some mutations as it’s spread around, as you can explore at NextStrain, so we’ve technically got choices as to which one to analyze. For this thread I’ll just stick to analyzing one version of the genome: Wuhan-Hu-1.
As a reminder, each A, G, C, and T in a genome is one of the four nucleotides: adenine, guanine, cytosine, and thymine. There are actually plenty of ways to engineer different unnatural base pair systems by adding artificial nucleotides, and these can even be integrated into transcription and translation, but for whatever reason, these four and not others are what life ultimately ended up with.
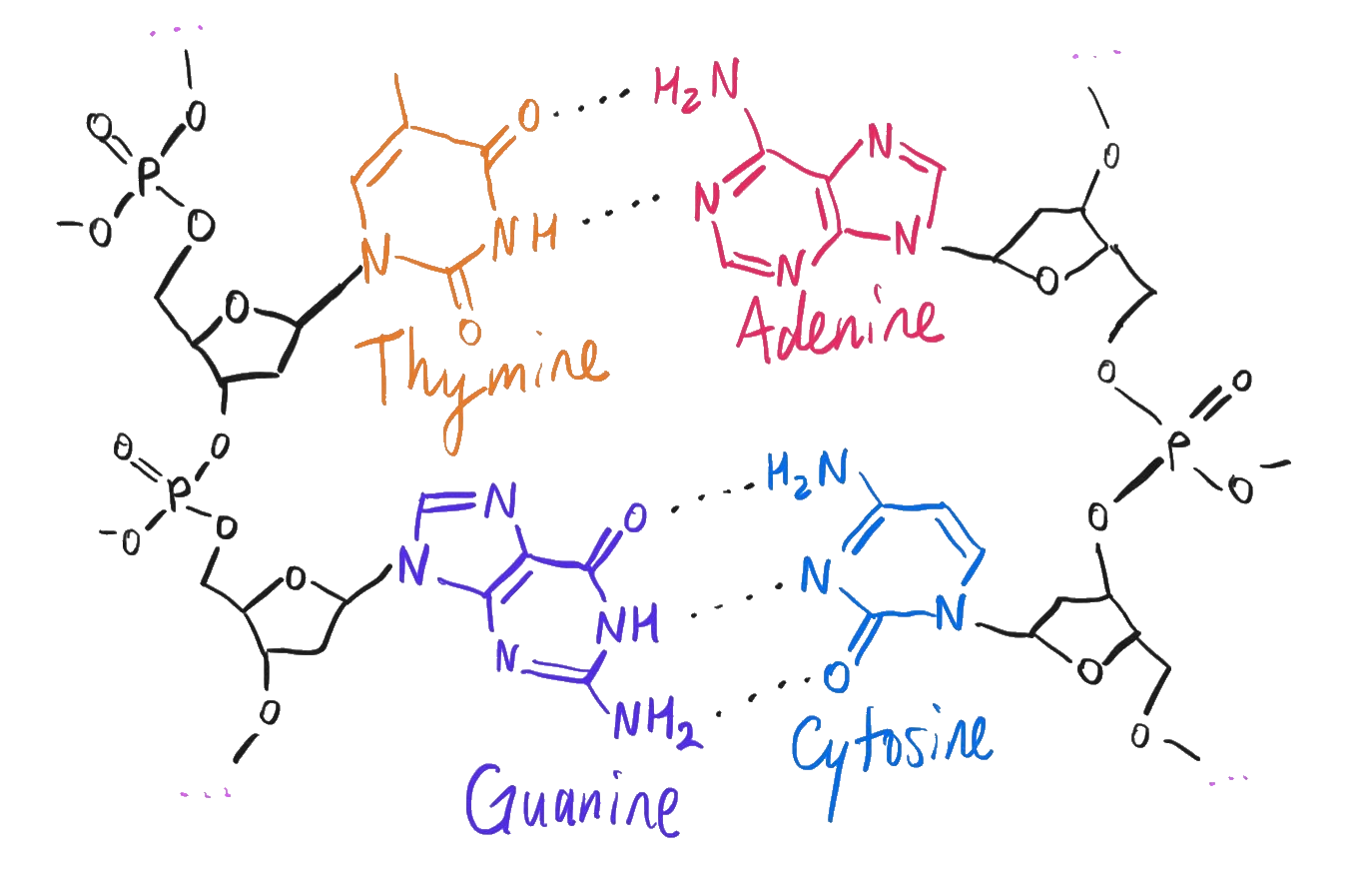
The four nucleotides in DNA.
The genome of Wuhan-Hu-1 is available from NCBI GenBank. Since SARS-CoV-2 is an RNA virus, each T in this string technically represents a U, for uracil, RNA’s information-equivalent of thymine. The genome sequence is therefore:
1 AUUAAAGGUU UAUACCUUCC CAGGUAACAA ACCAACCAAC UUUCGAUCUC UUGUAGAUCU
61 GUUCUCUAAA CGAACUUUAA AAUCUGUGUG GCUGUCACUC GGCUGCAUGC UUAGUGCACU
121 CACGCAGUAU AAUUAAUAAC UAAUUACUGU CGUUGACAGG ACACGAGUAA CUCGUCUAUC
...
29761 ACAGUGAACA AUGCUAGGGA GAGCUGCCUA UAUGGAAGAG CCCUAAUGUG UAAAAUUAAU
29821 UUUAGUAGUG CUAUCCCCAU GUGAUUUUAA UAGCUUCUUA GGAGAAUGAC AAAAAAAAAA
29881 AAAAAAAAAA AAAAAAAAAA AAA
Follow along with the genome »
That’s 29,903 nucleotides. Since there are only four possible nucleotides, we can estimate the information compression value of each nucleotide at approximately 2 bits; the virus’s genome therefore requires only 7.5 kilobytes to store. That’s roughly as much data, byte for byte, as there are characters in this essay up to this point!
Lay out those 29,903 nucleobases along a ribose-phosphate backbone, reading them left to right from the 5’ end to the 3’ end, and bam – if that single molecule* were teleported into a cell, that’s 100% chemically sufficient** to infect a person with the plague du jour.
*plus the 5’ cap, discussed below
**modulo viral load effects??
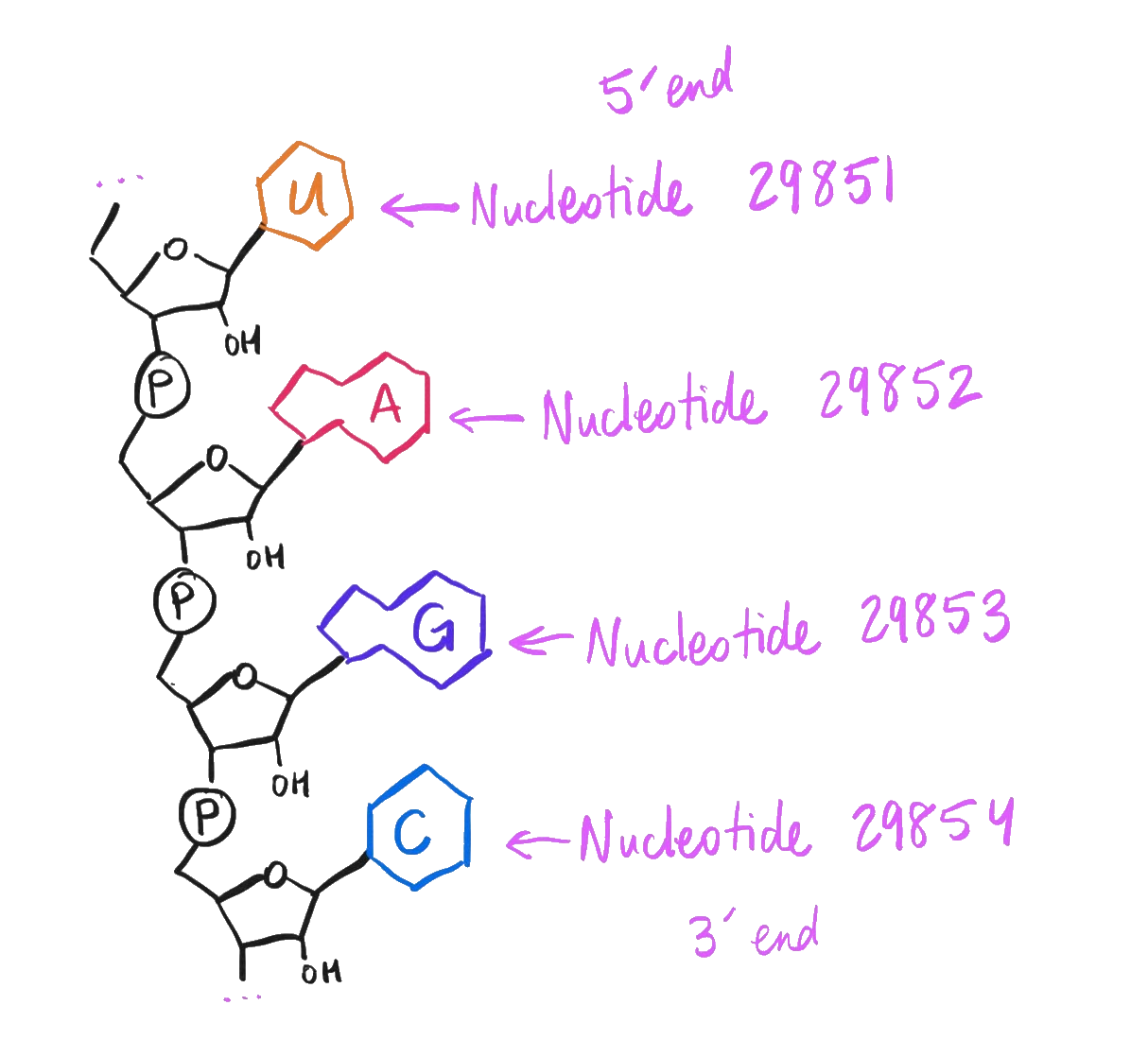
How to interpret the Wuhan-Hu-1 genome as a complete molecule.
Poly-A tail
First question, and perhaps the most obvious one to the naked eye – what’s with all the AAAAA at the end of the viral genome?
29821 ... ... AAAAAAAAAA
29881 AAAAAAAAAA AAAAAAAAAA AAA
Follow along with the genome »
It’s… yelling at us? Is it… suffering? Should we help?
Simple: It’s a 3’ poly-A tail! This long tail of adenosine monomers is extremely common in both our own cells and in RNA viruses.
Our own messenger RNA (mRNA) has a poly-A tail when it’s freshly produced in the nucleus so as to slow its degradation by the cell, allowing it to last long enough to be transcribed into protein. Naturally, if you’re a positive-strand RNA virus, you’re also going to want to last long enough to be transcribed into protein – so, you need the same feature, yourself.
Genome 0.11% explained. So far so good!
5’ cap
While we’re discussing chemical features of mRNA, note that the viral genome presumably must also have a 5’ cap – an extra 7-methylguanosine at the 5’ end of its RNA strand – just like mRNAs do.
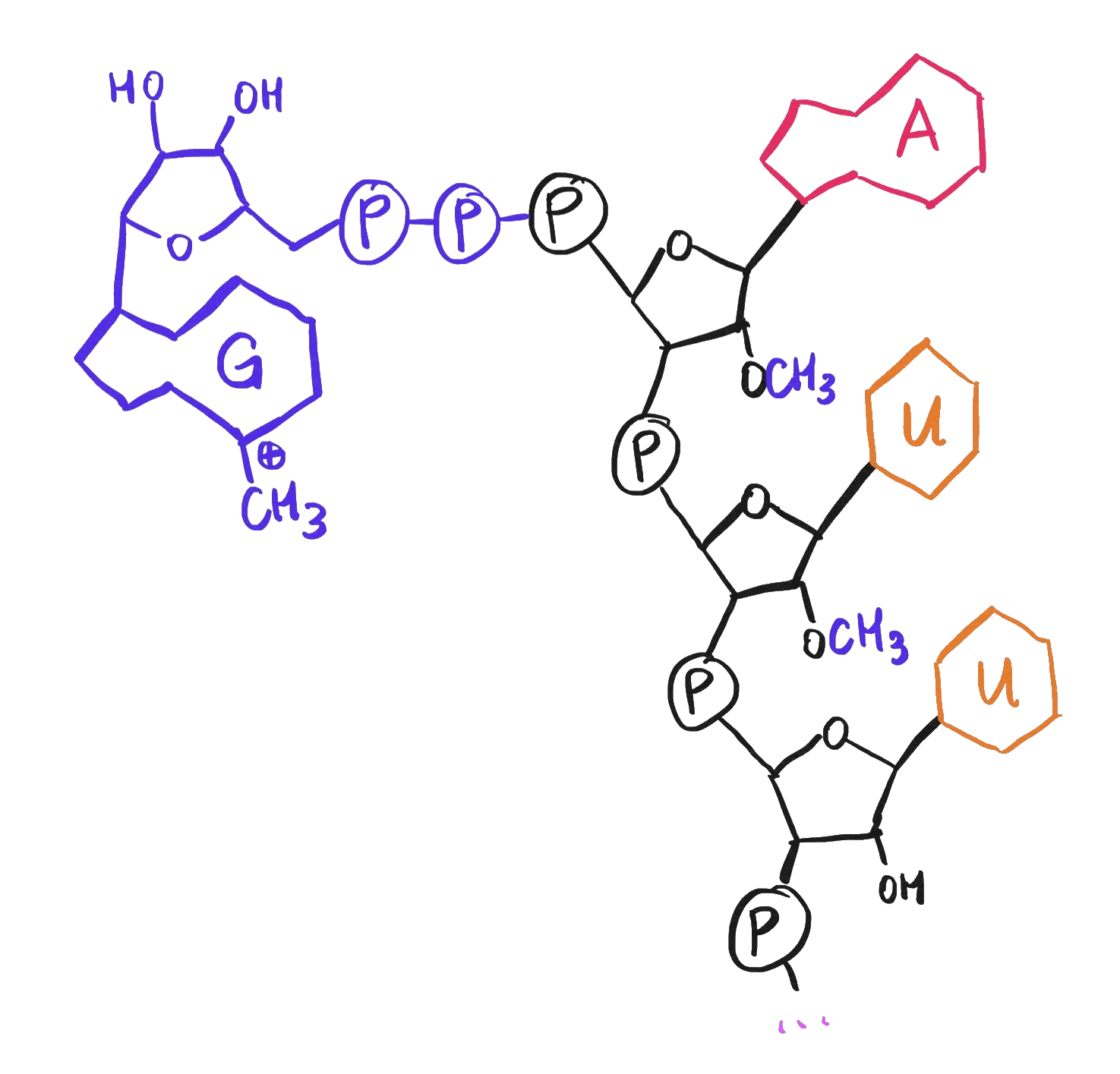
A 5' cap, consisting of a 7-methylguanosine as well as methylation of the first two ribose sugars.
The cap is not directly shown in the viral genome sequence or mentioned in NCBI GenBank, but it is referenced in multiple papers discussing coronaviral genomes:
Since 2003, the outbreak of severe acute respiratory syndrome coronavirus has drawn increased attention and stimulated numerous studies on the molecular virology of coronaviruses. Here, we review the current understanding of the mechanisms adopted by coronaviruses to produce the 5′-cap structure and methylation modification of viral genomic RNAs.

Coronaviruses possess a cap structure at the 5′ ends of viral genomic RNA and subgenomic RNAs, which is generated through consecutive methylations by virally encoded guanine-N7-methyltransferase (N7-MTase) and 2′-O-methyltransferase (2′-O-MTase). The coronaviral N7-MTase is unique for its physical linkage with an exoribonuclease (ExoN) harbored in nonstructural protein 14 (nsp14) of coronaviruses.
Here, we have reconstituted complete SARS-CoV mRNA cap methylation in vitro.
Like the poly-A tail, the 5’ cap helps the genome to be recognized and translated by ribosomes rather than destroyed by the cell’s immune response.
How does the virus even ensure that it receives a 5’ cap and a poly-A tail, not to mention its outer coat? Hopefully these questions will be resolved by our review of its genes… let’s move on to look at those!
Translation
Per the “Features” section of the genome, again from NCBI GenBank, here are the identifiable genes in this genome, in order:
Orf1ab(for orf1ab polyprotein)S(for surface glycoprotein)Orf3a(for orf3a protein)E(for envelope protein)M(for membrane glycoprotein)Orf6(for orf6 protein)Orf7a(for orf7a protein)Orf8(for orf8 protein)N(for nucleocapsid phosphoprotein)Orf10(for orf10 protein)
Let’s understand how these genes get translated into proteins.
Translation of Orf1ab
This is the first gene in the genome and it is also by far the longest, weighing in at 7,096 amino acids:
1 MESLVPGFNE KTHVQLSLPV LQVRDVLVRG FGDSVEEVLS EARQHLKDGT CGLVEVEKGV
61 LPQLEQPYVF IKRSDARTAP HGHVMVELVA ELEGIQYGRS GETLGVLVPH VGEIPVAYRK
121 VLLRKNGNKG AGGHSYGADL KSFDLGDELG TDPYEDFQEN WNTKHSSGVT RELMRELNGG
...
6961 LGGSVAIKIT EHSWNADLYK LMGHFAWWTA FVTNVNASSS EAFLIGCNYL GKPREQIDGY
7021 VMHANYIFWR NTNPIQLSSY SLFDMSKFPL KLRGTAVMSL KEGQINDMIL SLLSKGRLII
7081 RENNRVVISS DVLVNN
These letters are single-letter amino acid abbreviations.
It is quite long: this virus has 10 genes, and this single gene represents 71.2% of the viral genome.2 More on this polypeptide’s structure and function later, but first: how do the underlying nucleotides of the Orf1ab gene produce these particular amino acids?
A thermodynamic surprise: Ribosomal frameshift
The Orf1ab gene spans the range from nucleotide 266 to nucleotide 21,555, inclusive. Nucleotides in this GenBank data are unfortunately 1-indexed, not 0-indexed.
We can see at nucleotide 266 the signature AUG of a start codon, and at nucleotide 21,553 the UAA of an ochre stop codon. So far so good!
241 ... ...AUGGA GAGCCUUGUC CCUGGUUUCA ACGAGAAAAC
301 ACACGUCCAA CUCAGUUUGC CUGUUUUACA GGUUCGCGAC GUGCUCGUAC GUGGCUUUGG
361 AGACUCCGUG GAGGAGGUCU UAUCAGAGGC ACGUCAACAU CUUAAAGAUG GCACUUGUGG
...
21481 CUUAGUAAAG GUAGACUUAU AAUUAGAGAA AACAACAGAG UUGUUAUUUC UAGUGAUGUU
21541 CUUGUUAACA ACUAA... ...
Follow along with the genome »
However, confusingly, the length of this coding region is 21,555 - 265 = 21,290, which is not divisible by 3. Usually, 3 nucleotides = 1 amino acid, so a gene’s length is typically divisible by 3. What’s going on?
Note that in the GenBank data the gene is tagged ribosomal_slippage. Also note that in GenBank the gene’s region is notated as join(266..13468,13468..21555) instead of just 266..21555.
After some research, the answer here is that nucleotide 13,468 is actually used twice, thanks to a -1 ribosomal frameshift, a fascinating thermodynamic-biochemical quirk of certain viral genomes!
Per this article on ribosomal frameshifting in viruses:
Programmed ribosomal frameshifting is an alternate mechanism of translation to merge proteins encoded by two overlapping open reading frames. The frameshift occurs at low frequency and consists of ribosomes slipping by one base in either the 5’(-1) or 3’(+1) directions during translation. Some viruses contains both a +1 and a -1 ribosomal frameshift. […]
All cis-acting frameshift signals encoded in mRNAs are minimally composed of two functional elements: a heptanucleotide “slippery sequence” conforming to the general form
XXXYYYZ, followed by an RNA structural element, usually an H-type RNA pseudoknot, positioned an optimal number of nucleotides (5 to 9) downstream.
If we look around nucleotide 13,468, we do in fact find the heptanucleotide “slippery sequence” responsible: it’s UUUAAAC. That C is nucleotide 13,468 and it ends up getting transcribed twice.
13441 GUCAGCUGAU GCACAAUCGU UUUUAAACGG GUUUGCGGUG UAAGUGCAGC CCGUCUUACA
Follow along with the genome »
This frameshift gives us a total length of 21,291 nucleotides. Subtract 3 for the stop codon and then divide by 3, and we get a number which matches the reported protein sequence’s length: 7,096 amino acids. Hooray!
So, the math checks out. We now know what ribosomal_slippage and join(266..13468,13468..21555) mean, and we know how these 21,290 nucleotides become 7,096 amino acids. However, I still have two questions:
- What??
- How does ribosomal frameshifting even work??
Ribosomal frameshifting at the molecular level
Thermodynamic control of -1 programmed ribosomal frameshifting (Bock et al. 2019) explores how ribosomal frameshifting happens by performing free-energy molecular dynamics simulations. This paper also explains the structure and function of that heptanucleotide slippery sequence, stating:
Spontaneous ribosome slippage is a rare event that occurs, on average, once in 104–105 codons. This low spontaneous frameshifting increases dramatically on particular mRNAs that contain sequences for programmed ribosomal frameshifting (PRF). PRF requires a slippery sequence, which usually comprises a X XXY YYZ heptamer, where XXX and YYY are triplets of identical bases and Z is any nucleotide, which allows for cognate pairing of the P-site and A-site tRNAs in the 0-frame and −1-frame. The nature of the tRNAs bound to the slippery site codons is critical, including the modifications of nucleotides in the anticodon loop (i.e., at positions 34 and 37 of the tRNA).
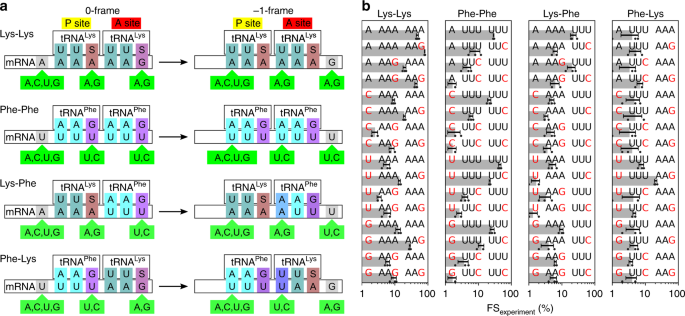
This paper goes on to analyze several heptanucleotide slippery sequences, drawing examples from the E. coli dnaX gene and explaining their thermodynamic characteristics. Per their breakdown, each example sequence depends on one or more of the following wobble pairings:
- The U·G wobble pair. Per Varani and McClain 2000, the U·G wobble pair “has comparable thermodynamic stability to Watson–Crick base pairs and is nearly isomorphic to them.”
- A·A and U·U mismatches.
- G·S and A·S pairs. Per Bock et al. 2019, E. coli “has a single tRNALys isoacceptor (anticodon 3’UUS5’) for decoding the two Lys codons, AAG and AAA,” where “S denotes the modified nucleotide mnm5s2U.”
For example, one of the heptanucleotide slippery sequences explained in Bock et al. is the heptanucleotide sequence UUUUAAG. When the ribosome reads this sequence, it initially parses it as ..U UUUPhe AAGLys, but then jolts -1 backwards into the ... UUUPhe UAALys reading frame. Despite normally being translated as a stop codon, that second UAA retains its attached tRNALys via the combination of a U·U mismatch and an A·S pair.

The UUUUAAG slippery sequence.
Unfortunately, UUUUAAG isn’t the sequence we’re interested in if we want to understand SARS-CoV-2! We need our particular heptanucleotide slippery sequence of interest, UUUAAAC, and despite this paper’s thoroughness and usefulness, none of its examples involve it. How can we be sure that UUUAAAC has the thermodynamic properties that it needs in order for SARS-CoV-2 to be able to produce a protein here?
After some investigation, I finally stumbled upon Mutational Analysis of the “Slippery-sequence” Component of a Coronavirus Ribosomal Frameshifting Signal (Brierley, Jenner, and Inglis 1992), a research paper which covers exactly this same heptanucleotide sequence (and in the context of coronaviruses, too), and even gives a helpful diagram!

![]()
The UUUAAAC slippery sequence.
The paper performs some experiments and confirms how the UUUAAAC slippery sequence works:
- First, a tRNALeu and a tRNAAsn bind to the
UUAandAAC. - After a -1 frameshift, those two tRNAs are now wobble-paired to
UUU(with a U·U mismatch) andAAA(with an A·G mismatch3). - Translation then proceeds as normal from there, with the next codon producing a tRNAArg.
I’m still a little weirded out by ribosomal frameshift, but satisfied.
Partial translation of Orf1ab
One last detail while we’re discussing the gene Orf1ab. Numerous papers I’ve read so far seem to allude to the fact that Orf1ab actually produces two protein products: one, its complete protein product (named pp1ab), and another, a partial translation (named pp1a) due to the ribosome falling off at the ribosomal frameshift instead of undergoing a frameshift event. That first half of the sequence itself can be called the gene Orf1a.
For example, from Graham et al. 2008 on the SARS coronavirus:
Translation of ORF1a results in a theoretical polyprotein of ∼500 kDa, while translation of ORF1ab results an ∼800 kDa polyprotein.
The ORF 1a and 1ab polyproteins are not detected during infection, since they are most likely processed co- and post-translationally into intermediate and mature proteins by proteinase activities in the nascent polyproteins.
Both of these genes produce polyproteins that actually get chopped up into smaller proteins before going on to carry out their function, so the possibility of premature termination of pp1a ends up being of little consequence except inasmuch as it partially reduces translation of pp1ab.
Translation of all the other genes
This concludes our analysis of the translation of the Orf1ab gene. Genome 71.31% explained so far!
In comparison, the remaining nine genes are fairly uneventful. They all start with a start codon (AUG), end with a stop codon (UAA, UGA, or UAG), and don’t try to do anything tricky in between.
| Gene | Start Nucleotide | End Nucleotide | Gene Length | Polypeptide Length |
|---|---|---|---|---|
S |
21563 | 25384 | 3822 | 1274 |
Orf3a |
25393 | 26220 | 828 | 276 |
E |
26245 | 26472 | 228 | 76 |
M |
26523 | 27191 | 669 | 223 |
Orf6 |
27202 | 27387 | 186 | 62 |
Orf7a |
27394 | 27759 | 366 | 122 |
Orf8 |
27894 | 28259 | 366 | 122 |
N |
28274 | 29533 | 1260 | 420 |
Orf10 |
29558 | 29674 | 117 | 39 |
By their powers combined, that explains 97.53% of the genome. If you take a look at the rest, you’ll see that there isn’t all that much left that’s not accounted for. With the two untranslated regions – there’s the 5’ UTR weighing in at 265 base pairs, and there’s the 3’ UTR (which includes the poly-A tail) weighing in at 229 base pairs. That covers 99.07% of the genome! The remaining 277 base pairs are scattered in the space between the ten genes.
I now feel basically confident that I know what the nucleotides get translated to!
Secondary structure
No discussion of the structure and function of a long, single-stranded piece of RNA is complete without a discussion on secondary structure.
Yes, RNA has secondary structure too – it’s not just for proteins! Just like double-helical DNA binds one strand to another, a single strand of RNA can bind to itself when regions have sufficiently complementary nucleotides, forming stems, hairloops, and yet more complex 3D structures.
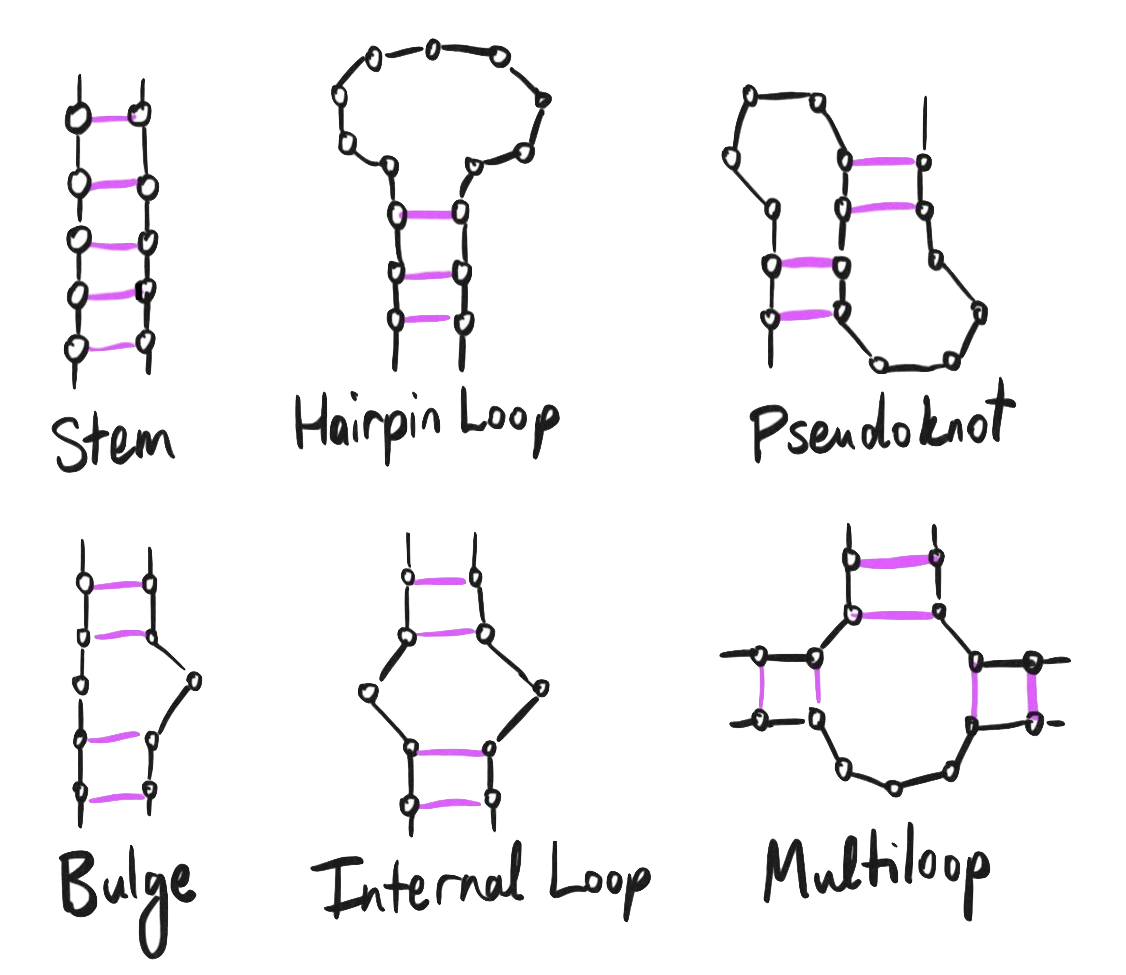
The pseudoknot may sound familiar – it’s mentioned back in our discussion on ribosomal frameshifting:
All cis-acting frameshift signals encoded in mRNAs are minimally composed of two functional elements: a heptanucleotide “slippery sequence” conforming to the general form
XXXYYYZ, followed by an RNA structural element, usually an H-type RNA pseudoknot, positioned an optimal number of nucleotides (5 to 9) downstream.
One of the more complex examples of RNA secondary structure, the pseudoknot was first discovered in the turnip yellow mosaic virus, which is itself another single-stranded positive-sense RNA virus just like the coronavirus.
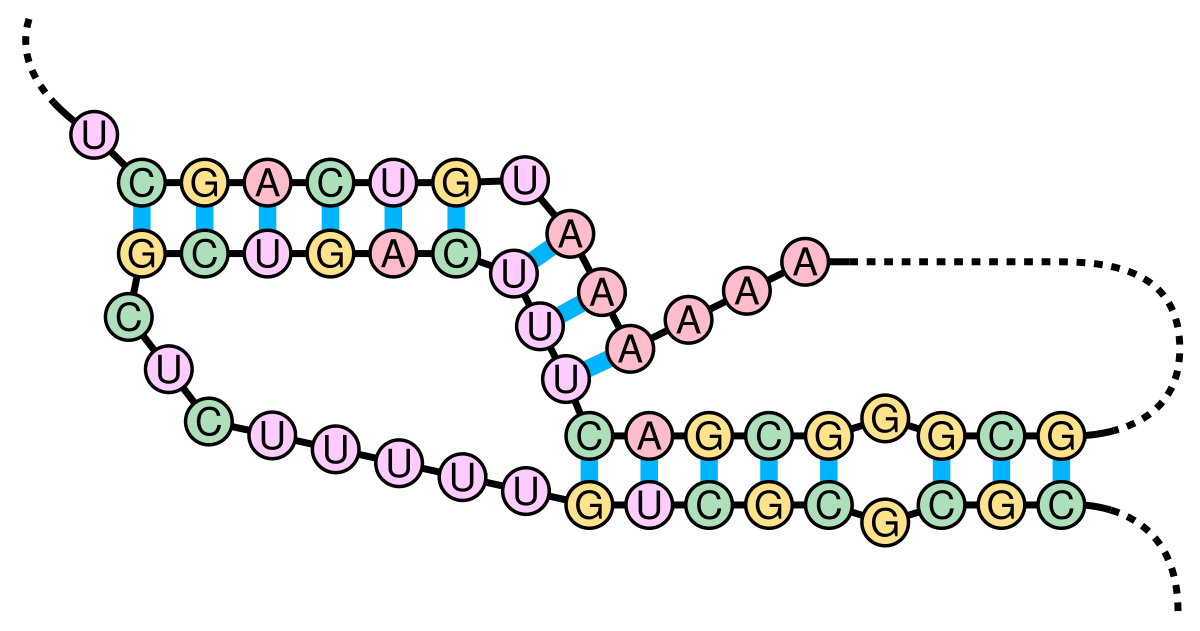
What’s an H-type RNA pseudoknot, though? According to Cao and Chen 2009,
An H-type pseudoknot is formed by base-pairing between a hairpin loop and the single-stranded region of the hairpin. The structure consists of two helix stems and two loops as well as a possible third loop/junction that connects the two helix stems.
Perhaps we can determine the secondary structure of the SARS-CoV-2 genome, or if not, at least determine the secondary structure for the pseudoknot, as that part serves an important regulatory function.
As we know from our study of ribosomal frameshifting, the pseudoknot should occur around 5 to 9 nucleotides downstream of the slippery sequence. Here’s a snippet of the surrounding genome sequence again:
13441 ...GU UUUUAAACGG GUUUGCGGUG UAAGUGCAGC CCGUCUUACA
13501 CCGUGCGGCA CAGGCACUAG UACUGAUGUC GUAUACAGGG CUUUUG...
Follow along with the genome »
For RNAs shorter than 4000 nucleotides, there exist some online tools such as RNAfold that can make predictions about the secondary structure of arbitrary RNAs; however, I’m unable to find any that can handle our 29,903-nucleotide minor behemoth of an RNA. So, that leaves me with searching through the existing literature to see what results exist for this particular big long RNA.
Luckily, RNA Genome Conservation and Secondary Structure in SARS-CoV-2 and SARS-Related Viruses (Rangan et al. 2020) gives an overview of some of the virus’s secondary structures, including the pseudoknot, as well as a thoughtful analysis of which structural elements have remained conserved as viruses in this family have evolved!
As depicted in that paper’s figure 4, here is how the region near the slippery sequence winds itself into an H-type pseudoknot:
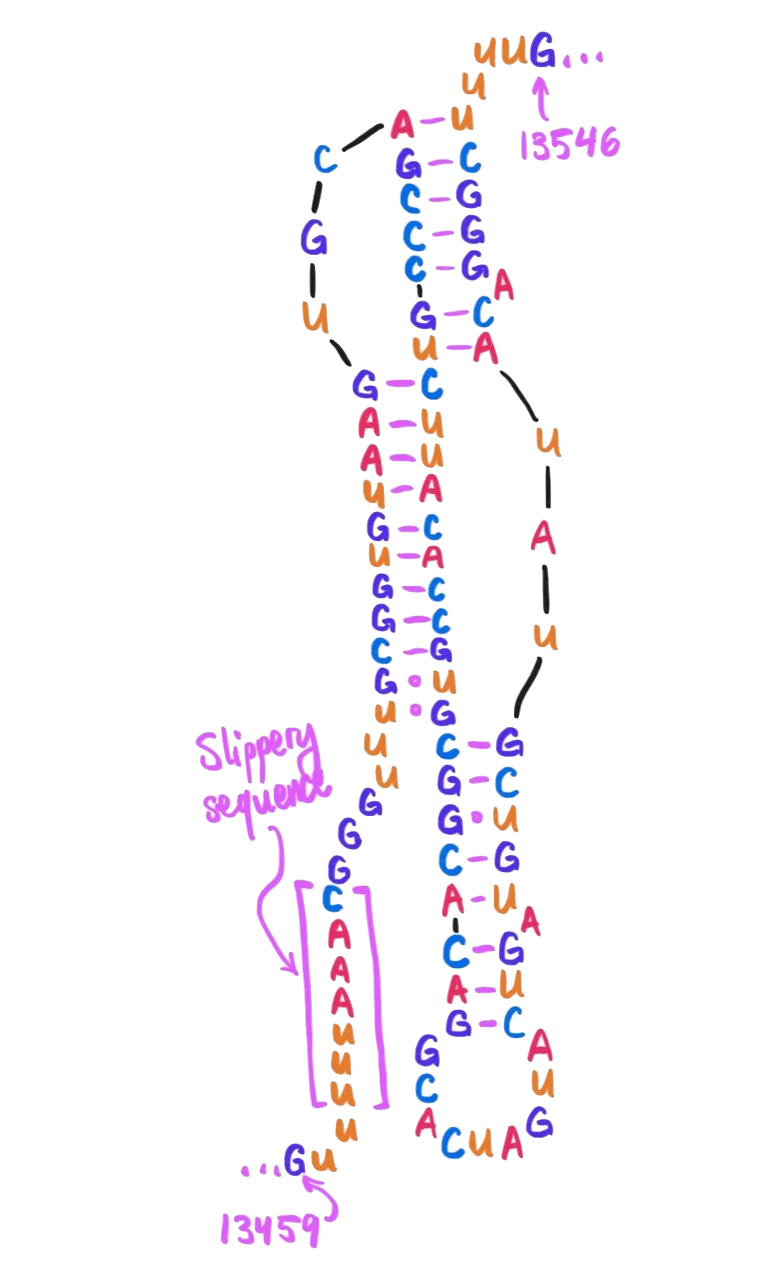
Starting 6 nucleotides after the heptanucleotide slippery sequence, nucleotides 13474 through 13542 together form a pseudoknot. (Here drawn schematically in 2D – in real life they wouldn't be so stretched out!)
Note the presence here of a few U·G wobble pairs, as mentioned in our earlier discussion on wobble pairings!
We can view this pseudoknot in 3D by converting the above secondary structure into dot-bracket notation, passing it into RNAComposer in order to extract a predicted 3D folding as a PDB file, and finally passing that into Web 3DNA to render a 3D image.
GUUUUUAAACGGGUUUGCGGUGUAAGUGCAGCCCGUCUUACACCGUGCGGCACAGGCACUAGUACUGAUGUCGUAUACAGGGCUUUUG
...............(((((((((((...[[[[[[[)))))))))))[[[[[[[[.........]]].]]]]]...]].]]]]]....
The pseudoknot in dot-bracket notation.

The pseudoknot, rendered in 3D.
For more information on secondary structure and how it can affect the life cycle and transmissibility of RNA viruses, as well as details about how secondary structure can be elucidated, I also recommend:
- RNA Structure—A Neglected Puppet Master for the Evolution of Virus and Host Immunity (Smyth et al. 2018)
- Viral RNAs Are Unusually Compact (Gopal et al. 2014)
- Visualizing the global secondary structure of a viral RNA genome with cryo-electron microscopy (Garmann et al. 2015)
- The influence of viral RNA secondary structure on interactions with innate host cell defences (Witteveldt et al. 2014)
Shoutouts
Thanks to Laura Vaughan for a thoughtful review of this piece and for help with RNA 3D structure visualization!
Shoutout also to Nabeel Qureshi who has worked on a similar endeavor, writing up an executable iPython notebook for understanding the full coronavirus genome.
As fate would have it, I know Ramya Rangan from our time as students of Jean Yang! It was really exciting to organically stumble upon a former colleague’s recent work, and especially to have so many of my questions about coronaviral secondary structure just answered immediately by that work.
Footnotes
-
Update: Alyssa Vance notes to me that targeting RdRP is actually the mechanism of action of remdesivir! Nabeel Qureshi adds that this is a property of favipiravir as well. From Wikipedia: “As an adenosine nucleoside triphosphate analog, the active metabolite of remdesivir interferes with the action of viral RNA-dependent RNA polymerase and evades proofreading by viral exoribonuclease, causing a decrease in viral RNA production.” ↩
-
A previous version of this essay claimed that all of orf1ab encodes for RdRP because I had seen that this protein product consumes monomers of RNA and catalyzes their polymerization. This is untrue! Nabeel Qureshi has pointed out to me that RdRP is only a small fraction of the orf1ab polyprotein. ↩
-
You might note that “A·G mismatch” is not among the canonical wobble pairing types that we had already discussed. I’m therefore a little skeptical of it; can it really be so thermodynamically favorable, especially given how A and G are such big unwieldy purines? Some quick research reveals that it’s at least possible, if uncommon. ↩